Что такое талассемия?
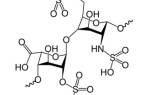
Красный кровяной пигмент, гемоглобин (Hb), является ключевым элементом эритроцитов. Он отвечает за перенос кислорода (О2) от легких к тканям и обратно, а также за утилизацию углекислого газа из тканей. Химическая структура молекулы гемоглобина напрямую связана с его функциями. Сложное строение Hb позволяет эффективно выполнять задачи, связанные с транспортировкой кислорода и обеспечением тканей питательными веществами. Эритроциты с аномальным гемоглобином теряют функциональность, что может привести к гипохромной анемии.
Гемоглобин человека представляет собой сложный белок, состоящий из четырех полипептидных цепей, связанных с гемом, содержащим двухвалентное железо (Fe2+). Гем играет важную роль в дыхательной функции крови. Глобин — это белковая часть молекулы Hb, которая взаимодействует с гемом. В его составе находятся по две альфа- и бета-цепи, каждая из которых состоит из аминокислотных остатков (в α-цепи — 141 аминокислот, в β-цепи — 146).
Дефектные гемоглобины возникают из-за замены аминокислотного остатка в одной из цепей глобина, что приводит к точечной мутации. Наибольшее количество замен происходит в β-цепях, поэтому β-талассемия является самой распространенной группой гемоглобинопатий, связанных с нарушением синтеза белкового компонента молекулы Hb. В то же время α-цепи имеют низкую вариабельность, что сказывается на распространенности α-талассемии.
Бета-талассемия, в отличие от альфа-талассемии, при которой теряются гены альфа-цепей, обычно не связана с делецией генов. Дефект возникает из-за формирования неполноценной м-РНК и бета-глобина, что снижает продукцию бета-цепей. Однако в результате создаются условия для избыточного накопления свободных α-цепей. Это приводит к распаду молодых (ядерных) форм эритроцитов в костном мозге, а зрелые красные клетки, попавшие в периферическую кровь, становятся уязвимыми. Неправильный гемоглобин в красных кровяных тельцах делает их менее устойчивыми и неэффективными. Из-за слабой мембраны красные кровяные клетки быстро разрушаются (гемолиз), что приводит к симптомам гемолитической анемии.
Талассемия представляет собой наследственное заболевание, связанное с нарушением синтеза гемоглобина, что приводит к анемии и другим осложнениям. Врачи отмечают, что это заболевание может проявляться в различных формах, включая альфа- и бета-талассемию, каждая из которых имеет свои особенности течения и степени тяжести. В зависимости от формы, пациенты могут испытывать различные симптомы, от легкой усталости до серьезных осложнений, таких как сердечная недостаточность.
Течение талассемии часто требует комплексного подхода к терапии. Врачи подчеркивают важность регулярного мониторинга состояния пациента и применения методов лечения, таких как переливания крови и хелатная терапия для удаления избытка железа. В некоторых случаях может быть рекомендовано проведение трансплантации костного мозга, что является единственным радикальным методом лечения. Специалисты также акцентируют внимание на необходимости генетического консультирования для семей, в которых есть случаи талассемии, чтобы предотвратить рождение детей с этим заболеванием.

Варианты и формы
Люди, унаследовавшие аномальный гемоглобин от обоих родителей, часто сталкиваются с тяжелыми последствиями, известными как большая талассемия (талассемия мажор) или болезнь Кули. Тем, кто получил этот «подарок» только от одного из родителей, повезло больше: они могут не осознавать наличие заболевания, если не возникнут неблагоприятные обстоятельства. Это указывает на то, что бета-талассемия имеет несколько форм:
- Гомозиготная β-талассемия, унаследованная от обоих родителей и названная в честь врача, который ее описал, болезнью Кули, характеризуется значительным увеличением уровня фетального гемоглобина (HbF) до 90%. Она проявляется у детей примерно к концу первого года жизни и сопровождается множеством симптомов.
- Гетерозиготная форма, известная как малая талассемия, возникает при наследовании заболевания только от одного из родителей. В этом случае признаки анемии, как правило, менее выражены, а иногда симптомы могут вовсе отсутствовать.
Тем не менее, такое разделение актуально лишь для классических случаев. На практике ситуация может быть более сложной. У некоторых пациентов с гомозиготной формой могут отсутствовать явные признаки тяжелого заболевания, и анемия не так выражена, чтобы требовать постоянных гемотрансфузий. Поэтому такую талассемию, несмотря на гомозиготность, не следует называть большой — ее классифицируют как промежуточную.
Кроме того, в рамках гомозиготной талассемии выделяют 3 степени тяжести:
- Гомозиготная талассемия, диагностируемая только у детей первого года жизни, так как они не могут прожить дольше из-за изменений в организме.
- Талассемия, хотя и менее тяжелая, не позволяет ребенку дожить до 8 лет.
- (назвать просто «легкой» не совсем корректно) — она дает возможность дожить до подросткового возраста и вступить во взрослую жизнь, но, к сожалению, может забрать эту жизнь в самом расцвете.
Некоторые случаи гетерозиготной бета-талассемии могут протекать более тяжело, чем ожидалось. Это несоответствие объясняется неспособностью красных кровяных клеток избавляться от избыточных альфа-цепей, хотя для гетерозиготной формы болезни характерна повышенная утилизация ненужных цепей. В связи с этим возникает необходимость классифицировать заболевание по формам в зависимости от степени выраженности клинических проявлений, независимо от гомо- или гетерозиготности. Выделяют:
- Большую талассемию;
- Промежуточную форму;
- Малую талассемию;
- Минимальный вариант.
Однако такое деление больше интересно специалистам, в то время как читателям, вероятно, интереснее узнать о ключевых признаках классических вариантов гомозиготных и гетерозиготных талассемий.
Эти патологические состояния, несмотря на общее название – β-талассемия, имеют значительные различия в анализах, симптомах, течении, лечении и прогнозе.
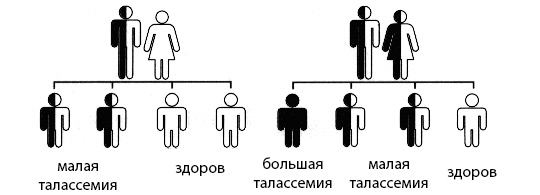
| Характеристика | Описание | Примечания |
|---|---|---|
| Понятие талассемии | Генетическое заболевание, характеризующееся снижением или отсутствием синтеза одного или нескольких глобиновых цепей гемоглобина. | Наследственное заболевание, передающееся по аутосомно-рецессивному типу. |
| Формы талассемии | α-талассемия (дефект в гене α-глобина) и β-талассемия (дефект в гене β-глобина). Внутри каждой формы существуют различные варианты тяжести, от бессимптомных до тяжелых. | Тяжесть зависит от количества дефектных генов. |
| Развитие талассемии | Мутации в генах, кодирующих α- или β-глобиновые цепи, приводят к дисбалансу в синтезе глобиновых цепей, что вызывает накопление избыточных цепей и повреждение эритроцитов. | Различные мутации приводят к различным клиническим проявлениям. |
| Течение β-талассемии (пример) | Может варьировать от бессимптомного носительства до тяжелой анемии (мажорная форма), требующей постоянных переливаний крови и хелатотерапии. | Интермедиарная форма занимает промежуточное положение по тяжести. |
| Течение α-талассемии (пример) | Может проявляться от бессимптомного носительства до тяжелой анемии (гидропс плода, смерть в перинатальном периоде). | Тяжесть зависит от количества дефектных генов. |
| Диагностика | Выявление мутаций в генах глобина, анализ крови (гемоглобин, гематокрит, ретикулоциты, электрофорез гемоглобина). | Пренатальная диагностика возможна. |
| Терапия β-талассемии (пример) | Регулярные переливания крови, хелатотерапия (удаление избытка железа), аллогенная трансплантация костного мозга, генная терапия (в стадии разработки). | Цель терапии – предотвращение осложнений и улучшение качества жизни. |
| Терапия α-талассемии (пример) | Лечение зависит от тяжести заболевания. Может включать переливания крови, трансфузионную поддержку, трансплантацию костного мозга. | Для тяжелых форм прогноз может быть неблагоприятным. |
| Осложнения | Железодефицитная анемия, гепатоспленомегалия, задержка роста, деформация костей, сердечная недостаточность, инфекции. | Осложнения связаны с накоплением железа и хронической анемией. |
| Профилактика | Генетическое консультирование, пренатальная диагностика. | Важно для семей с высоким риском рождения ребенка с талассемией. |
Гомозигота дает большую болезнь
Симптоматика бета-талассемии связана с нарушением баланса в производстве и изменением количества бета-цепей гемоглобина. Это приводит к неэффективному кроветворению. Красные кровяные клетки, не завершившие ядерную стадию, массово погибают в костном мозге. Зрелые эритроциты и, в меньшей степени, ретикулоциты преждевременно разрушаются в селезенке, что вызывает выраженные признаки анемии. В печени и селезенке также возникают новые очаги кроветворения. Интенсивное кроветворение нарушает нормальное развитие костной системы, приводя к деформациям и искажениям. Состояние выраженной гипоксии замедляет общее развитие организма ребенка. Такой тип течения болезни чаще наблюдается у пациентов с гомозиготной формой талассемии, чем у тех, кто страдает от гетерозиготной.
Талассемия — это наследственное заболевание крови, связанное с нарушением синтеза гемоглобина. Люди, страдающие от этой болезни, часто сталкиваются с анемией, усталостью и другими симптомами, связанными с недостатком кислорода в организме. Существует несколько форм талассемии, включая альфа- и бета-талассемию, каждая из которых имеет свои особенности и степень тяжести. Течение заболевания может варьироваться от легкой до тяжелой формы, требующей регулярного медицинского наблюдения.
Современные методы терапии включают регулярные переливания крови, которые помогают поддерживать уровень гемоглобина, а также препараты, способствующие выведению излишков железа из организма. В некоторых случаях может потребоваться пересадка костного мозга, что является радикальным, но эффективным методом лечения. Важно, чтобы пациенты получали своевременную диагностику и поддержку, что позволяет значительно улучшить качество их жизни.

Симптомы, обусловленные участием двух родителей
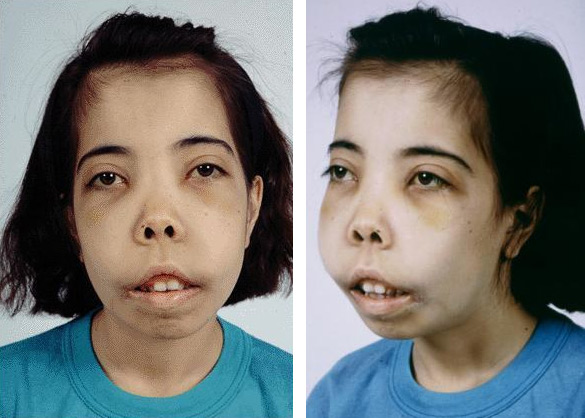
Большая талассемия проявляется с рождения, хотя в первые месяцы симптомы могут быть слабо выражены. К году, а в худшем случае к началу второго года жизни, клинические проявления становятся заметными:
- Обращает на себя внимание аномальная форма черепа (в виде башни или квадратной головы), увеличенная верхняя челюсть и характерные черты лица: монголоидный тип, маленькие глаза и плоская переносица (рентгенография выявляет патологические изменения).
- Является заметной бледность кожи, а также изменение ее оттенка – от сероватого до желтоватого.
- У детей рано наблюдается увеличение печени и селезенки, что связано с экстрамедуллярным (внекостным) кроветворением и накоплением гемосидерина в тканях (он образуется при распаде эритроцитов).
- Дети часто страдают от заболеваний из-за снижения иммунной защиты и отстают в физическом развитии. У тех, кто имеет более легкую форму гомозиготной бета-талассемии и доживает до подросткового и взрослого возраста, наблюдается задержка в развитии вторичных половых признаков.
- Лабораторный контроль подтверждает наличие клинических признаков тяжелой гемолитической анемии.
Анализы
Основным диагностическим критерием талассемии являются лабораторные исследования, которые позволяют охарактеризовать изменения в крови при этой патологии:
- Уровень гемоглобина снижается до 30-50 г/л.
- Цветной показатель показывает неблагоприятные результаты — 0,5 и ниже.
- Эритроциты в мазке имеют гипохромный вид, размеры клеток варьируются (выраженный анизоцитоз). Наблюдаются характерные для бета-талассемии (и мембранопатий) мишеневидные формы и базофильная зернистость.
- Осмотическая резистентность красных кровяных клеток повышена.
- Количество ретикулоцитов увеличивается.
- Биохимические показатели крови (билирубин за счет свободной фракции, сывороточное железо) также повышены. Избыточное накопление железа может привести к циррозу печени, развитию сахарного диабета и поражению сердечной мышцы.
Подтверждающим признаком диагноза является повышенное содержание фетального гемоглобина в эритроцитах (до 20-90%).

Лечение
Выраженное кислородное голодание тканей и активная работа кроветворной системы в костях (что не наблюдается у здоровых людей) являются показаниями для переливания полноценной донорской крови, содержащей эритроциты, способные обеспечить дыхание клеток. У пациентов с тяжелой формой талассемии гемотрансфузии с раннего возраста становятся основным методом лечения. Переливание крови не является обычным медикаментозным средством (как таблетки или растворы), поэтому к этому методу терапии следует подходить с особой осторожностью. Тем не менее:
- На первом этапе пациенту назначается интенсивный курс гемотрансфузий (в течение 2-3 недель он может получить до 10 переливаний, что позволяет повысить уровень гемоглобина до 120-140 г/л).
- На следующем этапе количество гемотрансфузий уменьшается, но стараются поддерживать уровень гемоглобина в пределах 90-100 г/л.
Такой подход к лечению позволяет:
- Значительно улучшить общее состояние пациента;
- Уменьшить негативное влияние заболевания на костную систему;
- Повысить иммунные функции организма;
- Предотвратить дальнейшее увеличение селезенки;
- Положительно сказаться на физическом развитии ребенка.
Несмотря на то, что переливание цельной крови практически не используется (вместо этого применяется эритроцитная масса), «неродная» биологическая среда несет риск осложнений:

- Пирогенные реакции (чаще всего возникают из-за нарушения технологии отмывания эритроцитов);
- Избыточное накопление гемосидерина (железосодержащего пигмента), что может привести к увеличению печени, сердечным заболеваниям и диабету.
Чтобы вывести из организма избыточное железо, пациентам назначают Десферал в виде внутримышечных инъекций (дозировка препарата определяется в зависимости от возраста и объема перелитой эритроцитной массы). Полезно сочетать Десферал с витамином С (аскорбиновой кислотой).
Значительное увеличение селезенки и сопутствующие изменения в крови (лейкопения, тромбоцитопения) могут стать основанием для удаления увеличивающегося органа (спленэктомии).
Устойчивое долгосрочное улучшение состояния у пациентов с гомозиготной β-талассемией возможно достичь с помощью трансплантации костного мозга, однако эта операция сопряжена с трудностями в подборе донора и может вызвать реакции после пересадки.
«Гетеро» подразумевает – «разный»
Гетерозиготная форма или малая талассемия возникает, когда один из родителей передает патологический ген потомству. При этой форме заболевания гетерозиготы обычно не проявляют всех признаков, поэтому болезнь может протекать бессимптомно или с минимальными проявлениями. Для малой талассемии характерны следующие клинические симптомы:
- Слабость, трудности с физической активностью и быстрая утомляемость;
- Легкая бледность кожи и незначительная желтушность склер и кожных покровов;
- Увеличение селезенки и, в некоторых случаях, печени, которое не всегда наблюдается.
Анализы при этой форме анемии показывают разнообразные показатели:
- Гипохромия эритроцитов, часто деформация клеток и изменение их размеров, а также базофильная зернистость;
- Уровень гемоглобина, как правило, снижен (90-100 г/л), но редко достигает критических значений (обычно не опускается ниже 70 г/л);
- Возможное значительное снижение ЦП (до 0,5);
- Общий билирубин (за счет несвязанной фракции) повышается не у всех пациентов (у 25% он остается в норме).
К важным диагностическим признакам малой талассемии относятся семейный анамнез (наличие заболевания у близких родственников) и определение уровня HbA2, который повышается до 4,5-9%, а также HbF, уровень которого у половины пациентов колеблется от 2,5 до 7%.
В большинстве случаев малая талассемия не требует лечения. Однако могут возникнуть проблемы из-за инфекционных процессов и беременности. В таких ситуациях пациентам назначают витамин В9 (фолиевая кислота) в суточной дозе 5-10 мг, так как в этих условиях потребление этого витамина организмом увеличивается.
Напоследок – несколько слов о снижении продукции α-цепей
Продукция α-цепей красного кровяного пигмента регулируется двумя парами генов, что приводит к различным формам заболевания:
 Читайте также:
Читайте также:

- При полном отсутствии альфа-цепей в эмбриональный период развития ребенка красный пигмент крови не образуется. В таких случаях результат, как правило, трагичен — это водянка головного мозга и смерть ребенка.
- Если нарушаются функциональные возможности одного или двух генов из этих пар, симптомы заболевания начинают напоминать клинические проявления β-талассемии. Однако лабораторные показатели, касающиеся HbF и HbA2, значительно отличаются, и уровень этих гемоглобинов не увеличивается.
Лечение α-талассемии в целом не отличается от терапевтических подходов, применяемых для гетерозиготной формы β-талассемии.
Видео: талассемия, программа “Жить здорово”
Вопрос-ответ
Что такое талассемия?
Талассемии — группа наследственных заболеваний кроветворной системы, которые характеризуются нарушением синтеза гемоглобина. Поэтому основным проявлением болезни является анемия. Как известно, нормальный человеческий гемоглобин HbA состоит из четырех белковых цепей двух разных видов: две α-цепи и две β-цепи.
Что применяют для лечения талассемии?
В лечении талассемии применяются гемотрансфузии, терапия десфералом, спленэктомия, трансплантация костного мозга.
Какие виды талассемии бывают?
Существует два основных типа: альфа-талассемия и бета-талассемия.
Как лечить носителя талассемии?
Трансплантация костного мозга и стволовых клеток от совместимого родственного донора — единственный метод лечения талассемии. Совместимость означает, что клетки донора имеют одинаковые типы белков, называемых человеческими лейкоцитарными антигенами (HLA), на поверхности реципиента трансплантата.
Советы
СОВЕТ №1
Регулярно проходите медицинские обследования, особенно если у вас в семье есть случаи талассемии. Раннее выявление может помочь в своевременной диагностике и лечении заболевания.
СОВЕТ №2
Обратите внимание на свое питание. Убедитесь, что ваш рацион богат железом и витаминами, особенно витамином B12 и фолиевой кислотой, которые могут помочь поддерживать здоровье крови.
СОВЕТ №3
Если у вас диагностирована талассемия, обсудите с врачом возможность генетического консультирования, особенно если вы планируете завести детей. Это поможет понять риски передачи заболевания.
СОВЕТ №4
Поддерживайте связь с группами поддержки и сообществами людей с талассемией. Это может помочь вам обмениваться опытом и получать эмоциональную поддержку от тех, кто сталкивается с аналогичными проблемами.
-
Читайте также: